
Синдром Марфана
Содержание
Синдром Марфана

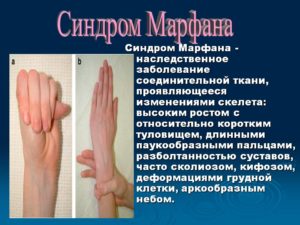
Синдром Марфана — дифференцированная форма врожденной соединительнотканной недостаточности, характеризующаяся разнообразными проявлениями скелетной, сердечно-сосудистой и глазной патологии.
У больных с синдромом Марфана отмечаются гигантизм, долихостеномелия и арахнодактилия, аневризмы аорты, миопия, эктопия хрусталика, деформация грудины, кифосколиоз, плоскостопие, протрузия вертлужной впадины, эктазия твердой мозговой оболочки.
Диагноз синдрома Марфана основан на семейном анамнезе, результатах функционального, офтальмологического, рентгенологического и генетического исследований. Лечение при синдроме Марфана включает консервативную и хирургическую коррекцию сердечно-сосудистых нарушений, поражений скелета и органа зрения.
Синдром Марфана — системное недоразвитие соединительной ткани в эмбриональном и постнатальном периодах, обусловленное структурными дефектами коллагена и сопровождающееся преимущественным поражением опорно-двигательного аппарата, глаз, сердечно-сосудистой системы.
Синдром Марфана — одна из наиболее распространенных наследственных коллагенопатий синдромального характера.
Частота встречаемости синдрома Марфана в популяции невысока: по данным различных авторов составляет 1 случай на 10000-20000 человек, без расовой и половой детерминированности.
Причины синдрома Марфана
Синдром Марфана относится к врожденным аномалиям, наследуемым по аутосомно-доминантному типу, с выраженным плейотропизмом, варьирующей экспрессивностью и высокой пенетрантностью.
В основе синдрома Марфана лежат мутации в гене N1, отвечающем за синтез фибриллина – важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани.
Аномалия и дефицит фибриллина при синдроме Марфана приводят к нарушению формирования волокнистых структур, потере прочности и упругости соединительной ткани, невозможности выдерживать физиологические нагрузки.
Гистологическим изменениям в большей степени подвержены стенки сосудов эластического типа и связочный аппарат (в первую очередь, аорта и цинновая связка глаза, содержащие наибольшее количество фибриллина).Широкий фенотипический спектр синдрома Марфана (от легких форм, трудно отличимых от нормы до тяжелых, быстропрогрессирующих) объясняется разнообразием мутаций в гене N1 (более 1000 видов), а также присутствием мутаций в других генах (например, в гене трансформирующего фактора роста — TGR-2). При генетическом исследовании в 75% случаев синдрома Марфана выявляется семейный тип наследования, в остальных — первичная мутация. Риск рождения ребенка с синдромом Марфана возрастает с увеличением возраста отца (особенно после 35 лет).
В зависимости от количества пораженных систем выделяют несколько форм синдрома Марфана:
- стертую — со слабо выраженными изменениями в 1-2-х системах
- выраженную — со слабо выраженными изменениями в 3-х системах; выраженными изменениями хотя бы в 1-ой системе; выраженными изменениями в 2-3-х и более системах.
Степень тяжести изменений при синдроме Марфана может быть легкой, средней и тяжелой. По характеру течения дифференцируют прогрессирующий и стабильный синдром Марфана.
Симптомы синдрома Марфана
Синдром Марфана характеризуется сочетанным поражением скелета, глаз, сердечно-сосудистой и нервной систем; многообразием проявлений, варьированием сроков появления первых признаков заболевания; хроническим прогредиентным течением.
Больные синдромом Марфана, как правило, отличаются высоким ростом, относительно коротким туловищем с непропорционально длинными тонкими конечностями (долихостеномелией) и удлиненными паукообразными пальцами (арахнодактилией); астеническим телосложением со слаборазвитой подкожной клетчаткой и мышечной гипотонией; длинным и узким лицевым скелетом (долихоцефалией); наличием высокого аркообразного неба и нарушения прикуса (прогнатии). Средняя длина тела при рождении у мальчиков с синдромом Марфана составляет 53 см, окончательный рост – 191 см; у девочек — соответственно 52,5 см и 175 см.
При синдроме Марфана отмечаются нарушение функции суставов (гипермобильность); деформация грудной клетки (воронкообразная или килевидная форма), деформация позвоночника (сколиоз, кифоз, кифосколиоз, подвывихи и вывихи шейного отдела, спондилолистез), а также плоскостопие и протрузия вертлужной впадины.
Сердечно-сосудистая патология, доминирующая в клинической картине синдрома Марфана и часто определяющая его исход, проявляется дефектами структуры стенок сосудов эластического типа, особенно аорты и крупных ветвей легочной артерии, пороками развития клапанного аппарата и перегородок сердца.
Изменения аорты у больных синдромом Марфана характеризуются прогрессирующим расширением ее восходящей части и клапанного кольца (дилатацией, аннулоаортальной эктазией) и аневризмами; поражение митрального клапана — миксоматозной дегенерацией створок, патологическим удлинением и разрывом створочных хорд, обызвествлением клапанного кольца. У плода с синдромом Марфана возможно формирование врожденных пороков сердца — коарктации аорты, стеноза легочной артерии, ДМПП и ДМЖП. Органические и функциональные изменения сердца и сосудов у больных синдромом Марфана часто сопровождаются нарушением ритма (наджелудочковой и желудочковой тахикардией, фибрилляцией предсердий) и развитием инфекционного эндокардита.
Самая неблагоприятная неонатальная форма синдрома Марфана проявляется в классическом варианте уже при рождении, приводит к прогрессирующей сердечной недостаточности и летальному исходу на первом году жизни ребенка.
Для большинства случаев синдрома Марфана характерна патология органа зрения, включающая близорукость, вывих/подвывих (эктопию) хрусталика, уплощение и увеличение размера роговицы, гипоплазию радужной оболочки и цилиарной мышцы, косоглазие, изменение калибра сосудов сетчатки. Эктопия хрусталика при синдроме Марфана имеет двухсторонний характер, часто развивается в возрасте до 4-х лет и устойчиво прогрессирует, ухудшая зрительную функцию.
При синдроме Марфана наблюдается поражение других систем и органов: нервной (эктазия твердой мозговой оболочки, в т. ч.пояснично-крестцовое менингоцеле), бронхолегочной (спонтанный пневмоторакс, эмфизема легких, дыхательная недостаточность), кожи и мягких тканей (атрофические стрии), рецидивирующие паховые и бедренные грыжи, вывихи и разрывы связок, а также эктопия почек, опущение мочевого пузыря и матки, варикозное расширение вен и др.
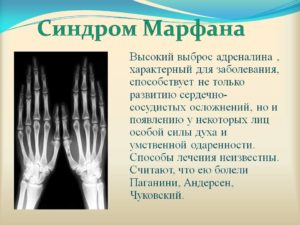
Характерный для синдрома Марфана высокий выброс адреналина может способствовать постоянному нервному возбуждению, гиперактивности, а иногда развитию неординарных способностей и умственной одаренности.
Диагноз синдрома Марфана основывается на семейном анамнезе, наличии у больного типичных диагностических признаков по результатам физикального осмотра, ЭКГ и ЭхоКГ, офтальмологического и рентгенологического обследования, молекулярно-генетического анализа и лабораторных исследований.
За диагностические критерии синдрома Марфана берутся характерные изменения в различных системах и органах; главными (большими) из них считаются: дилатация корня/расслоение восходящей части аорты, эктопия хрусталика и эктазия твердой мозговой оболочки; килевидная/воронкообразная деформация грудной клетки, требующая хирургического лечения; отношение длины верхнего сегмента тела к нижнему 1,05; сколиоз (> 20˚) или спондилолистез; ограничение разгибания в локтевом суставе (
Также применяются фенотипические диагностические тесты, определяющие соотношение кисть/рост (при синдроме Марфана > 11%); длину среднего пальца (> 10 см); индекс телосложения Варги – (масса тела, г/(рост, см)x2 – возраст, годы/100, должно быть
ЭКГ при синдроме Марфана позволяет определить нарушение ритма сердца, выраженную гипертрофию миокарда; ЭхоКГ — обнаружить клапанную регургитацию, увеличение размеров левого желудочка, пролапс митрального клапана, разрывы хорд, дилатацию аорты. На рентгенографии грудной клетки можно увидеть расширение корня и дуги аорты, увеличение размеров сердца; на КТ и МРТ сердца и сосудов — выявить дилатацию и аневризмы аорты.
Аортография показана при подозрении на аневризму и расслоение аорты. Наличие эктопии хрусталика уточняют с помощью биомикроскопии и офтальмоскопии; протрузию вертлужной впадины устанавливают методом рентгенографии тазобедренных суставов; эктазию твердой мозговой оболочки – МРТ позвоночника.
При синдроме Марфана определяется возрастание (в 2 раза и более) почечной экскреции метаболитов соединительной ткани: глюкозоаминогликанов и их фракций. Метод прямого автоматического секвенирования ДНК позволяет провести генетическую идентификацию мутаций в гене N1.Необходима дифференциальная диагностика с заболеваниями, внешне напоминающими синдром Марфана: гомоцистинурией, врожденной контрактурной арахнодактилией (синдромом Билса), наследственной артроофтальмопатией (синдромом Стиклера), MASS-синдромом, синдромами Элерса-Данлоса, Лойса-Дитца, Шпринцена–Голдберга, семейной эктопией хрусталика и др.
Лечение синдрома Марфана
Лечение и дальнейшее наблюдение пациентов с синдромом Марфана должно осуществляться группой специалистов: офтальмологом, кардиологом, кардиохирургом, ортопедом, генетиком, терапевтом.
Лечение больных с синдромом Марфана направлено на профилактику прогрессирования заболевания и развития осложнений, в первую очередь в сердечно-сосудистой системе. При диаметре аорты до 4 см назначаются β-адреноблокаторы, антагонисты кальция или ингибиторы АПФ.
Хирургическое лечение проводится при недостаточности клапанов сердца, пролапсе митрального клапана, значительном расширении (>5 см) восходящей части и расслоении аорты. Реконструктивные операции на аорте при синдроме Марфана, имеют высокий процент послеоперационной 5-ти и 10-ти летней выживаемости.
При необходимости выполняют протезирование митрального клапана. У беременных с синдромом Марфана и выраженной сердечно-сосудистой патологией проводят досрочное оперативное родоразрешение путем кесарева сечения.
С целью профилактика инфекционного эндокардита и тромбозов после операционных вмешательств назначаются антибиотики и антикоагулянты.
При синдроме Марфана проводится коррекция зрения с помощью подбора очков и контактных линз, при необходимости – лазерное или хирургическое лечение катаракты, глаукомы, удаление смещенного хрусталика с имплантацией искусственного.
При выраженных скелетных нарушениях может потребоваться хирургическая стабилизация позвоночника, торакопластика, эндопротезирование тазобедренных суставов.Применяются также патогенетическая коллагеннормализующая терапия, метаболическая и витаминотерапия.
Прогноз
Прогноз жизни больных с синдромом Марфана определяется, в первую очередь, степенью сердечно-сосудистых изменений, а также поражений скелета и глаз.
Имеется высокий риск осложненного течения, снижения продолжительности жизни (90-95% не доживают до 40-50 лет) и внезапной смерти.
Своевременная кардиохирургическая коррекция при синдроме Марфана позволяет значительно увеличить продолжительность (до 60-70 лет) и улучшить качество жизни больных.
Больные синдромом Марфана должны находиться под постоянным врачебным наблюдением и регулярно проходить диагностическое обследование.
При синдроме Марфана показан низкий или средний уровень физической активности, исключающий занятия контактными видами спорта, спортивные соревнования, изометрические нагрузки, подводное плавание.
Женщинам детородного возраста с синдромом Марфана необходимо пройти медико-генетическое консультирование.
Источник: https://www.krasotaimedicina.ru/diseases/children/marfan-syndrome
Синдром Марфана
Приветствую, дорогие читатели. Сегодня хотел бы поговорить про высоких людей с Синдромом Марфана, для которых высокий рост – это не просто особенность организма, а симптом серьезной болезни, требующей к себе постоянного внимания.
Синдром Марфана — заболевание наследственного типа, при котором поражается соединительная ткань с вовлечением в процесс скелетно-мышечной системы и глаз. Установлено, что причиной патологии является мутация гена фибриллина N1. Заболевание полиморфно — может протекать с разной выраженностью клинической картины, и характеризуется появлением все новых типов мутации в генах.
Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который вперые представил описание 5-летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.
Распространенность синдрома — 1 случай на 10000 человек. Риск рождения ребенка с синдромом Марфана повышается после достижения отцом возраста 35 лет и достигает 50% при наличии патологии у одного из родителей. Врожденная аномалия наследуется по аутосомно-доминантному типу. В ее основе лежит дефект важнейшего гена, отвечающего за синтез коллагена.
Во время внутриутробного развития происходит нарушение формирования волокон соединительной ткани, утеря ими прочности, в результате чего волокна не способны выдерживать естественные нагрузки. Поэтому наибольшие атипичные изменения претерпевают крупные сосуды, клапаны сердца, связки глаза, твердое небо, скелет и мышцы.
Без адекватной терапии продолжительность жизни людей с синдромом Марфана не более 40 лет. Терапия позволяет увеличить этот срок вдвое и более.
История заболевания
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани.
К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа.
Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности).Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.
Симптомы синдрома Марфана
Многообразие вариантов генетической мутации обуславливает различные формы течения болезни. Нередко они малозаметны, иногда приводят к инвалидизации человека в раннем возрасте. Частый признак синдрома Марфана — высокий рост (до 200 см.
), при этом туловище непропорционально короткое, а конечности удлиненные и тонкие.
Пальцы у больных длинные, паукообразные (арахнодактилия) Из-за недоразвития подкожной клетчатки и мышечной дистрофии страдающие синдромом Марфана имеют астеническое телосложение.
Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
— гиперподвижность суставов;— аномалии строения тазобедренного сустава;— кифоз, сколиоз;— вывихи шейного сегмента позвоночника;— деформация грудной клетки;— плоскостопие;— глубокая посадка глаз;— уменьшенная нижняя челюсть, нарушение роста зубов;— высокое нёбо;— атрофические «растяжки» на коже;
— паховые грыжи, частые разрывы связок.
Более серьезные изменения при синдроме Марфана протекают в организме. Самые тяжелые из них развиваются со стороны сердца и сосудов и могут привести к смерти ребенка еще на первом году жизни. Среди них:
— дефекты ветвей легочной артерии, аорты (расширения, аневризмы, расслоения);— пороки сердца (чаще — поражения клапанов);
— стенозы артерий.
Подобные нарушения вызывают тахикардию, мерцательную аритмию вплоть до фибрилляции предсердий или развития сердечной недостаточности.
Со стороны глаз наблюдается выраженная миопия, вывих хрусталика, аномалии развития роговицы, уменьшение в размерах радужки, косоглазие, патологии сосудистой стенки сетчатки. При прогрессирующем вывихе хрусталика или при отслойке сетчатки уже в раннем возрасте больные могут полностью потерять зрение.
Со стороны нервной системы при синдроме Марфана происходит растяжение твердой мозговой оболочки и выбухание ликвора в костные дефекты в пояснично-крестцовом отделе позвоночника (дуральная эктазия).
Легкие страдают гораздо реже, так как незначительные нарушения их работы не оказывают влияния на дыхательную функцию. Но в отдельных случаях снижение эластичности альвеол может привести к спонтанному пневмотораксу, развитию дыхательной недостаточности.
Прочими симптомами патологии могут быть эктопия почек, деформации мочевого пузыря, половых органов.
Лечение и профилактика осложнений
Специфической терапии заболевания не существует: изменить гены еще до рождения ребенка невозможно. Лечение только симптоматическое и зависит от тех изменений в организме, которые развиваются у больного синдромом Марфана. Некоторые осложнения патологии можно успешно корректировать, другие — устранять оперативным путем.
Пациент должен наблюдаться у группы специалистов — офтальмолога, невролога, кардиолога, ортопеда, хирурга. Основное направление терапии — поддержка функций сердца и сосудов.
Методы лечения:
— прием препаратов (адреноблокаторы, антиаритмические лекарства, антикоагулянты и т.д.);
— хирургия пороков сердца (дисфункции клапанов, расширения, расслоения легочной артерии), аорты, протезирование клапанного аппарата.
Нормализация зрения проводится при помощи коррекции миопии (ношение очков, линз), лечения катаракты, глаукомы, имплантации искусственного хрусталика.
При поражении суставов и позвоночника проводится оперативное лечение (протезирование, пластика суставов, устранение межпозвоночных грыж), выправление кифоза, сколиоза при помощи тракции, мануальной терапии. Из медикаментозных средств используются миорелаксанты, витамины группы В. Также применяется физиолечение, занятия ЛФК.При поражении легких часто требуется хирургическое вмешательство (дренирование их полости).
Беременность больными синдромом Марфана должна строго планироваться и развиваться под контролем группы врачей, специализирующихся на лечении людей с подобными патологиями. Родоразрешение — только при помощи кесарева сечения.
Еще до наступления беременности желательно обследоваться на предмет возможного прогрессирования расслойки аорты и, по возможности, провести операцию по замене части сосуда.
Консультация генетика позволит рассчитать примерный риск по передаче заболевания по наследству.
Известные люди с синдромом Марфана
Хоть синдром Марфана – очень редкое заболевание, есть немало знаменитостей, больных синдромом Марфан: Фло Хайман (призер Олимпийских игр по волейболу), Джон Тавенер (композитор), Джоуи Рамон (музыкант), Лесли Хорнби (фотомодель и певица) и другие.
Среди исторических личностей, известных во всем мире, с синдромом Марфана можно выделить:
Музыкант-скрипач Никколо Паганини. Поскольку Паганини умер еще до описания синдрома, данные о его заболевании исследуются по сохранившимся изображениям и дневнику лечащего врача. Скрипач имел характерную деформацию пальцев, высокий рост и худобу, непропорциональное развитие конечностей, впалую грудь, мышечную слабость.
Писатель Ганс Кристиан Андерсен. Имел угловатое лицо, был очень худым и длинноруким, рано заполучил проблемы со зрением.
Президент Америки Авраам Линкольн. Кроме всех внешних признаков синдрома Марфана у Линкольна наблюдались ревматические боли, «разболтанность» суставов, но, в то же время — хорошая физическая выносливость.
Писатель Корней Чуковский. Наличие большого непропорционального носа, длинных конечностей не помешало Чуковскому стать одним из лучших творцов современности и доктором филологии.
Осама Бен Ладен. «Террорист №1» мира имел высокий рост и малый вес и большие проблемы с суставами и позвоночником, а также вытянутый череп и слишком узкое лицо.
Источники и ссылки:
1. Причины высокого роста — статья, в которой рассмотрены основные причины высокого роста человека.
2. Синдром Марфана в википедии — о синдроме Марфана в свободной энциклопедии.
3. Русскоговорящее сообщество людей с синдромом Марфана — форум о синдроме Марфана, общение, обсуждение болезни.
4. Международный фонд синдрома Марфана — фонд помощи больным с синдромом Марфана.
5. Группа вконтакте для больных синдромом Марфана.
Источник: http://tall.by/2014/sindrom-marfana/
Болезнь Марфана: причины, симптомы и лечение
Болезнь (синдром) Марфана – это наследственная патология соединительной ткани. Поскольку соединительная ткань является основой различных органов человека, то и проявления заболевания весьма разнообразны.
Самыми типичными являются аномалии сердечно-сосудистой системы, опорно-двигательного аппарата и глаз. Диагностика синдрома Марфана требует использования самых разных методов. Лечение включает в себя медикаментозные и оперативные способы, причем наблюдаться больной должен у нескольких врачей.
Из этой статьи Вы сможете узнать о причинах, симптомах, диагностике и вариантах лечения данного заболевания.
Синдром Марфана формируется еще во внутриутробном периоде, ребенок рождается уже с имеющимися нарушениями. Это, пожалуй, наиболее часто встречающаяся наследственная патология соединительной ткани. Независимо от пола и расовой принадлежности, частота выявления болезни Марфана колеблется от 1: 5 000 до 1:10 000 населения (по разным данным).
Причины
Основой заболевания служит генетический дефект: мутация гена, ответственного за синтез белка соединительной ткани фибриллина. Ген расположен на 15-й хромосоме.
Наследуется заболевание по аутосомно-доминантному типу. Аутосомное означает, что оно не связано с полом, а доминантное – то, что мутация всегда проявляется. Степень проявления (распространенность нарушений и их выраженность) может варьироваться в различных пределах, что связано с генетическими особенностями.
Фибриллин придает соединительной ткани эластичность и растяжимость. Нарушение его строения приводит к утрате прочности и упругости соединительной ткани, и она перестает служить прочным каркасом. В первую очередь изменения касаются стенок сосудов, связочного аппарата. Даже небольшие физиологические нагрузки становятся запредельными для организма, соединительная ткань их не выдерживает.
Все случаи синдрома Марфана с точки зрения причины подразделяют на:
- семейные: составляют 75%, это передающаяся из поколения в поколение мутация гена;
- случайные (спорадические): 25%, это впервые возникшая мутация гена в роду, где ранее не было подобной патологии.
Симптомы
Клинических проявлений заболевания весьма много. Их рассматривают с точки зрения поражения отдельных органов и систем:
- опорно-двигательного аппарата;
- сердечно-сосудистой системы;
- органа зрения;
- нервной системы;
- бронхолегочной системы;
- кожи, мягких тканей;
- других органов.
Опорно-двигательный аппарат
Для больных с патологией соединительной ткани характерна гипермобильность суставов.
Больные с синдромом Марфана обычно имеют высокий рост, худощавы из-за недоразвития подкожно-жировой клетчатки. Туловище при этом кажется коротким, а конечности – непропорционально длинными.
Размах рук больше роста на 5% и более. Череп вытянутый (долихоцефалический), пальцы длинные паукообразные (арахнодактилия).
Лицо вытянутое, узкое, небо высокое аркообразное (готическое), уши большие, нижняя челюсть выступает вперед (неправильный прикус), зубы растут неправильно, глаза глубоко посажены.
Деформация скелета заключается в наличии воронкообразной или килевидной грудной клетки, любых изменений в оси позвоночника (избыточный лордоз, кифозы, кифосколиозы, сколиозы), подвывихов позвонков (особенно в шейном отделе), спондилолистеза (смещение вышележащего позвонка по отношению к нижележащему).Характерно плоскостопие различных степеней выраженности, разболтанность суставов, избыточные движения в них (например, переразгибание), протрузия вертлужной впадины (углубление с глубоким погружением головки бедренной кости в области тазобедренного сустава).
Обычно все это сопровождается снижением мышечного тонуса.
Сердечно-сосудистая система
Изменения в этой системе часто являются наиболее тяжелыми, требующими коррекции в первую очередь, поскольку могут иметь угрожающий жизни характер.
Чаще всего синдром Марфана проявляется наличием пороков развития сердца и крупных сосудов (в частности, аорты).
Дефекты строения сердца заключаются в нарушении перегородок сердца и его клапанов (пролапс митрального клапана, дефект межжелудочковой или межпредсердной перегородки, патологическое удлинение хорд).
Нарушение строения стенки аорты приводит к развитию ее дилятации (расширению) и расслоению стенок (расслаивающая аневризма аорты). Из врожденных пороков крупных сосудов могут наблюдаться коарктация аорты, стеноз (сужение) легочной артерии.
Патологическое строение сердца и сосудов сопровождается развитием сердечной недостаточности, нарушением сердечного ритма (наджелудочковая и желудочковая тахикардии, фибрилляция предсердий и других), что весьма опасно для жизни больного. Присоединение инфекционных осложнений чревато развитием инфекционного эндокардита.
Орган зрения
Симптомы поражения глаз очень специфичны для данного заболевания. Поражение опорно-двигательного аппарата, сердечно-сосудистой системы и глаз составляют типичную триаду симптомов при болезни Марфана.
Больные синдромом Марфана часто имеют голубые склеры. Зрение плохое ввиду развития близорукости высокой степени, длина глазного яблока увеличена. Зрачки могут быть не симметричными, то есть разными по размеру.
Однако этим поражение органа зрения не ограничивается. Развиваются косоглазие, вывих или подвывих хрусталика, недоразвитие радужки и ресничных мышц, уплощение роговицы. Возможна отслойка сетчатки с развитием слепоты.
Нервная система
Поражение нервной системы в основном связано с патологией строения стенки сосудов. Это приводит к нарушению кровообращения, развитию ишемических или геморрагических инсультов (чаще – субарахноидальных кровоизлияний).
Возможны частые обмороки. Из аномалий возможны эктазия твердой мозговой оболочки, в частности пояснично-крестцовое менингоцеле (выпячивание оболочек мозга под кожу с образованием кармана через дефект в позвонках).
Возможны отклонения в психомоторном развитии, причем как в сторону задержки, так и в сторону превышения показателей. Многие люди с синдромом Марфана имеют высокие показатели интеллекта (выше, чем среднестатистический показатель IQ в популяции).Еще одним симптомом при болезни Марфана могут служить гипофизарные расстройства с повышением содержания адреналина в крови, развитием акромегалии и несахарного диабета.
Бронхолегочная система
Поражение бронхов и легких заключается в развитии спонтанных пневмотораксов, дыхательной недостаточности, эмфиземы легких, апикальных булл (пузырьковидные образования в верхушках легких). Все эти изменения становятся результатом нарушения ангиоархитектоники легких из-за патологии соединительной ткани, повышенной растяжимости и пониженной эластичности легочной ткани.
Поражение кожи и мягких тканей
Часто больные с синдромом Марфана имеют атрофические стрии на коже: волнистые полосы на коже разной ширины и цвета (от белого до красно-фиолетового). Малое количество подкожно жировой клетчатки становится причиной малого веса таких больных.
Больные с синдромом Марфана страдают рецидивирующими паховыми и бедренными грыжами.
Другие органы
Патология соединительной ткани становится причиной опущения почек, мочевого пузыря и матки, варикозного расширения вен, приводит к образованию длинного и со слабой перистальтикой кишечника (что сопровождается запорами).
Диагностика
Врач может заподозрить наличие синдрома Марфана у больного по результатам осмотра и обследования.
Диагностика синдрома Марфана требует учета анамнестических данных (в том числе и наследственного анамнеза), данных осмотра больного, а также проведения разнообразных дополнительных методов исследования, позволяющих обнаружить изменения в сердце, сосудах, головном мозге, легких, глазах.
Для этого могут понадобиться ЭКГ, ЭхоКГ (УЗИ сердца), рентгенография грудной клетки и тазобедренных суставов, КТ или МРТ головного и спинного мозга, сердца и сосудов, офтальмоскопия, аортография и другие методы. Перечень исследований зависит от наличия симптомов у конкретного больного.
Существуют диагностические критерии этого заболевания: большие и малые. Определенное сочетание больших и малых критериев и позволяет подтвердить диагноз.
К большим критериям, например, относят подвывих хрусталика, эктазию твердой мозговой оболочки, килевидную или воронкообразную грудную клетку, плоскостопие, протрузию вертлужной впадины и другие.
К малым критериям относят избыточную подвижность в суставах, готическое небо, деформацию черепа, спонтанный пневмоторакс и другие.
Окончательный диагноз выставляется после применения молекулярно-генетического метода и установления мутации гена, ответственного за синтез фибриллина.
Лечение
Лечение синдрома Марфана всегда требует одновременного участия нескольких специалистов: кардиолога, кардиохирурга, офтальмолога, ортопеда-травматолога, терапевта.
Спектр лечебных процедур охватывает консервативные и оперативные способы лечения. Консервативные меры направлены на профилактику осложнений и поддержание нормального функционирования органов и систем, а оперативные предполагают коррекцию имеющихся анатомических изменений с целью предотвратить выраженное нарушение функций или даже угрозу для жизни больного.
Хирургические методы лечения:
- реконструктивные операции на аорте (при значительном, более 5 см, расширении восходящей части аорты и расслоении ее стенки);
- протезирование клапанов сердца;
- удаление измененного хрусталика с заменой его на искусственный;
- пластика позвоночника при выраженном сколиозе;
- эндопротезирование тазобедренных суставов;
- пластика грудной клетки (в последние годы отрицается ее целесообразность).
Больному необходим подбор очков или контактных линз, иногда возможна лазерная коррекция зрения.
Медикаментозное лечение имеет патогенетическую и симптоматическую направленность.
Больному с метаболической целью назначают большие дозы витамина С (1-3 г в сутки), препараты с глюкозаминосульфатами и хондроитинсульфатами (Терафлекс, Структум, Хондроксид, Глюкозамин, Аминоартрин, Эльбона, Юниум), Карнитина хлорид 20% раствор, Янтарную кислоту по 100-200 мг 2 раза в день, препараты магния (например, Магне В6), поливитаминно-минеральные комплексы (с кальцием, магнием, цинком, медью). Прием этих веществ направлен на нормализацию обмена веществ, укрепление соединительной ткани.
Для лечения сердечно-сосудистых нарушений часто используют β-адреноблокаторы (Пропранолол, Обзидан, Атенолол), блокаторы кальция (Нифедипин, Амлодипин, Лекоптин), ингибиторы ангиотензинпревращающего фермента (Лизиноприл, Периндоприл, Эналаприл), антиаритмические препараты (при нарушении ритма сердца). При развитии инфекционного эндокардита показаны антибиотики. После оперативных вмешательств может понадобиться антикоагулянтная терапия для снижения свертываемости крови (Фраксипарин, Клексан).
В целом подбор метода лечения и ассортимент применяемых лекарственных средств очень индивидуальны. Все зависит от спектра клинических симптомов и выраженности нарушений у конкретного больного.Больным с синдромом Марфана показана лечебная физкультура, но в строго дозированном количестве, чтобы занятия приносили пользу сердечно-сосудистой и опорно-двигательной системам, а не вред. Нельзя заниматься контактными и игровыми видами спорта (баскетбол, футбол), рекомендовано плавание.
Прогноз
Заболевание неизлечимо, но постоянное динамическое наблюдение за больным, хирургическая коррекция сердечно-сосудистых, суставных и офтальмологических нарушений позволяют снизить риск для жизни, улучшить ее качество, позволить заниматься своей профессией. Некоторые больные не доживают до 40 лет, что связано с сердечно-сосудистыми осложнениями, в других случаях продолжительность жизни составляет 70 лет (особенно после проведения кардиохирургических операций).
Таким образом, синдром или болезнь Марфана – это наследственное заболевание, течение которого во многом зависит от тщательности медицинского наблюдения и своевременности оказания медицинской помощи. Обычно лечение требует привлечения многих специалистов, но такой комплексный подход позволяет избежать множества осложнений и повысить качество жизни больного.
Источник: https://doctor-neurologist.ru/bolezn-marfana-prichiny-simptomy-i-lechenie
Синдром Марфана
Синдром Марфана относится к моногенным болезням соединительной ткани — группе различных по происхождению нозологических форм, которые объединяют наследственные нарушения обмена соединительной ткани.
Большая часть этой патологии обусловлена нарушением ферментных систем, контролирующих синтез структурных белков из которых происходит синтез соединительной ткани.
Почти все эти болезни, входящие в состав синдрома, приводят к тяжелым инвалидизирующим расстройствам.
Синдром Марфана впервые описан Вильямсом в 1876 г. Свое название заболевание получило от французского педиатра Марфана, наблюдавшего девочку с характерным симптомокомплексом болезни 20 лет спустя.
Существует интересный факт, что первая девушка модель — Лесли Хорнби, которая послужила прототипом образа всех моделей, имела синдром Марфана.
Так, установлено, что ряд всемирно известных людей страдали синдромом Марфана, среди них следует упомянуть президента США А. Линкольна и великого скрипача Паганини.
Лесли Хорнби (Твигги)
Симптомы синдрома Марфана
Клиническая картина заболевания характеризуется поражением многих жизненно важных органов и систем: опорно-двигательного аппарата, сердечно-сосудистой системы, органов дыхания и зрения, ЦНС.
Так, среди конституциональных особенностей и нарушений скелета наиболее часто встречаются долихопластический (астенический) тип, высокий рост (как правило, выше 180см) при выраженном дефиците массы тела (обычно ниже 50 кг.
), арахнодактилия («паучьи» пальцы) кистей и стоп, кифосколиоз, воронкообразная или килевидная деформации грудной клетки, плоскостопие, узкий лицевой скелет, «готическое» небо.
Не обязательно у одного человека встречаются все эти признаки, это лишь наиболее распространееные
Арахнодактилия
Внешний вид больного с синдромом Марфана
Типичны также антимонголоидный разрез глаз, «крупный» нос, большие низкорасположенные ушные раковины, «птичье» выражение лица. У новорожденных из перечисленных особенностей скелета, как правило, выявляются только долихопластический тип и арахнодактилия. Остальные симптомы формируются в более поздние периоды развития (обычно в течение первых семи лет жизни ребенка).
Поражение сердца и сосудов — один из кардинальных признаков синдрома Марфана. Наиболее типичны среди них — пролапс митрального клапана и аневризма аорты. Сердечно-сосудистые нарушения регистрируются уже на первом-втором годах жизни ребенка, при этом отмечается постепенное увеличение диаметра аорты, достигающее критических размеров (до 6 см и более), чаще в возрасте от 16 до 45 лет.
Грозным осложнением аневризмы аорты является расслоение ее стенок, которое может быстро прогрессировать, захватывая всю длину аорты и отходящие от нее сосуды. Такие осложнения, как правило, заканчиваются летально.
Бронхолегочная система также вовлекается в патологический процесс при синдроме Марфана. Предпосылкой для этого являются механическое сдавление дыхательных путей при деформациях грудной клетки и изменения соединительнотканных структур легочной ткани.
Нарушения органов дыхания в виде спонтанного пневмоторакса, легочной эмфиземы, инфаркта легкого встречаются с частотой от 10 до 75%.Наряду с этим, имеются сведения о врожденном недоразвитии одной из долей легкого, поликистозе легких, врожденной буллезной эмфиземе, двусторонних бронхоэктазах.
К наиболее типичной патологии органа зрения при синдроме Марфана относится вывих и подвывих хрусталика (вследствие слабости цинновой связки). Как правило, эта патология сочетается с миопией или гиперметропией высокой степени.
Подвывих хрусталиков диагностируется обычно на 1-5 годах жизни, а иногда даже в 7 лет при оформлении ребенка в школу. Реже встречаются вторичная глаукома, катаракта, отслойка сетчатки.
Эти изменения чаще выявляются у больных более старшего возраста 15- 40 лет.
IQ (коэффициент интеллектуального развития) у большинства детей с синдромом Марфана обычно соответствует норме – 85-115 единиц. Встречаются лица с очень высоким интеллектом, у которых IQ превышает верхнюю границу нормы -115 ед.
Однако может иметь место определенное своеобразие психических процессов, которое проявляется в неравномерной интеллектуальной деятельности, а также в личностных особенностях больных (раздражительности, плаксивости, завышенной самооценке).
Для всех детей с синдромом Марфана характерна низкая переносимость физической нагрузки, которая нередко сопровождается болями в мышцах.
Возможны также периодические приступы мигренеподобной головной боли, возникающей, как правило, на фоне или после эмоционально-физических нагрузок.Эти признаки заболевания в сочетании со слабостью, гипотонией и гипоплазией мышечной ткани, а также нарушениями показателей физического развития служат свидетельством изменения функции митохондрий (нарушением процессов клеточной биоэнергетики).
Диагностика синдрома Марфана
Диагностика синдрома Марфана базируется на генеалогических данных (составление и анализ родословных) и анализе морфофенотипа, который включает изучение физического, нервно-психического развития детей и состояния физического развития больных проводится с помощью перцентильных шкал Стюарта.
О пропорциональности или гармоничности отдельных частей тела судят путем использования индекса Дю Ранта-Лайнера, который вычисляется по формуле А/В х 100, где А — отношение фактической массы тела к 50 перцентилю массы, соответствующей росту больного, а В — отношение фактической длины тела к 50 перцентилю роста соответствующего возраста. Индекс количественно отражает вариации физического развития. При этом показатель 89 и ниже соответствует высокому росту при дефиците массы тела, показатели — 110-119 — избыточной массе тела, свыше 120 – ожирению. Для детей, свыше 120 – ожирению. Для детей с синдромом Марфана индекс Дю Ранта-Лайнера, как правило, составляет 51-81.
По резолюции совещания, посвященного синдрому Марфана, для постановки диагноза необходимо наличие минимум одного из пяти основных симптомов заболевания (вывих хрусталиков, аневризма аорты, арахнодактилия, деформация грудины, кифосколиоз) и двух дополнительных (миопия, пролапс митрального клапана, умеренная гиперподвижность суставов, высокий рост, плоскостопие, стрии, пневмоторакс).
Установлено что в 90% всех случаев синдрома Марфана трудностей в постановке правильного диагноза, как правило, не возникает. Однако в 10% — диагностика затруднена.
В подобных ситуациях особенно необходимо чрезвычайно тщательное обследование максимально большего числа родственников больного.
Программа обследования для таких семей должна обязательно включать консультативные, осмотры окулиста, кардиолога и проведение эхокардиографии.
В диагностике синдрома Марфана широко используются также результаты рентгено-функциональных методов исследования.
Так, для оценки арахнодактилии используют показатели метакарпального индекса (отношение длины к ширине второй-пятой метакарпальных костей), вычисляемого по рентгенограмме правой кисти.У больных с синдромом Марфана наблюдается увеличение этого показателя до 8,0-11,0 при норме 6,4-7,9.
Характер и степень тяжести сердечно сосудистой патологии оценивают по данным эхокардиографии, ЭКГ, холтеровскому мониторированию. Анализ состояния бронхолегочной системы проводят по результатам исследования функции внешнего дыхания.
У подавляющего числа детей с синдромом Марфана регистрируются изменения этих показателей проявляющиеся в нарушении механики дыхания, вздутии легочной ткани, неравномерности распределения в легких вдыхаемого воздуха гиперкапнии.
У детей с синдромом Марфана обнаруживается снижение репарационной способности ДНК-лимфоцитов, что необходимо принимать во внимание при рентгенологическом обследовании выборе профессии и места жительства больных.
При синдроме Марфана имеет место увеличение (в два раза и более) выведения с мочой гпикозаминогликанов и их фракций, при этом особенно резко возрастает почечная экскреция хондроитин-4-6-сульфатов и в меньшей степени — гиалоурановый кислоты и гепарин-сульфата.
Дифференциальную диагностику проводят со сходными синдромами и генетическими заболеваниями. В качестве критерий сравнения выступает ряд признаков проявления различных заболеваний.
| Признаки | Различные синдромы со схожими симптомами | |||
| Марфана | Билса | Стиклера | Вейла-Марчезани | |
| Астеническое телосложение | + | + | + | — |
| Арахнодактилия | + | + | + | — |
| Деформация грудной клетки | +/- | +/- | — | — |
| Аневризма аорты | + | — | — | — |
| Подвывих хрусталика | + | — | — | +/- |
| Аутосомно-доминантный тип наследования | + | + | + | — |
Лечение синдрома Марфана
Детям с синдромом Марфана показана комплексная терапия, включающая широкий спектр лекарственных препаратов — средства, влияющие на сердечнососудистую систему, стимуляторы ЦНС, энерготропные препараты и антиоксиданты по схеме «Комплекс терапевтических воздействий, применяемый для лечения больных с синдромом Марфана»
Бета-адреноблокаторы — обзидан, атенолол — 10 мг/сут, длительность 6-12 и более месяцев.
Энерготропные и антиоксидантные препараты:1. рибоксин — 1 таб. (0,2) 2 раза в день в течение 1 мес, 3 курса в год;2. витамины В1 В2- 10 мг/сут, 10 дней ежемесячно;3. аскорбиновая кислота до 500 мг/сут, в течение 1 мес, 3-4 курса в год;4.
токоферол (вит Е) до 100 мг/сут, в течение 3-4-месячных курсов в год;5. элькар — 200-400 мг/сут, 3 мес, 2-3 курса в год;6. димефосфон — 30 мг/кг — 1 мес, 3-4 курса в год, • коэнзимQ10 — 30 мг 2-3 раза в сут, 3 мес, 2-3 курса в год,7. лимонтар — 5 мг/кг/сут, 10 дней, 4 курса в год,8.
ноотропные препараты пирацетам -200-400 мг 2 раза в сут в течение 2 мес, 3 курса в год
Наряду с медикаментозными средствами, детям с синдромом Марфана необходим также комплекс дополнительных лечебных воздействий, включающий магнитотерапию на суставы (курс 10 сеансов, 3 курса в год), электросон (курс 10 сеансов — дважды в год), лечебную физкультуру с преимущественным воздействием на опорно-двигательный аппарат (курс 14 дней, 4 курса в год), санаторий для больных с нарушениями функций костей и суставов или сердечно-сосудистой системы (курс лечения 24 дня — 1 раз в год) но показаниям, проводится оперативное лечение торакопластика, аневризмэктомия, пластика аорты, экстракция хрусталика, тонзилэктомия и аденотомия. Осуществляется также регулярная санация (не менее двух раз в год) хронических очагов инфекции полости рта и зубов.
Под воздействием комплексной терапии у 78-80% детей с синдромом Марфана отмечаются улучшение или стабилизация основного патологического процесса.
Клиническими критериями эффективности проводимого лечения служат повышение толерантности к физическим нагрузкам, нарастание мышечной силы, стабилизация диаметра аорты (по данным эхокардиографии) и тенденция к нормализации показателей функции внешнего дыхания, улучшение мелкой моторики, повышение эмоционального тонуса, увеличение объема произвольной памяти и концентрации внимания, повышение школьной успеваемости. Положительная биохимическая динамика проявляется в снижении уровня молочной и пировиноградной кислот.
При наблюдении за больными с синдромом Марфана необходимо выполнять следующие требования к режиму труда, отдыха и реабилитации:
1. детям с синдромом Марфана разрешаются занятия физкультурой только по ослабленной программе (спецгруппы и групы ЛФК);2. категорически запрещаются занятия в спортивных секциях, участие в соревнованиях, сельскохозяйственных работах, походах на длительные дистанции по пересеченной и горной местности, ношение тяжестей (не более 3 кг);3.
категорически запрещены специальности, связанные с профессиональной вредностью: контакты с химическими веществами, лаками, красками, работа в условиях высоких температур и воздействия радиации, а также профессии, сопряженные с вибрацией, требующие высокой остроты зрения, больших физических и эмоциональных затрат;4.
при выборе места жительства больным противопоказаны жаркий климат и зоны повышенной радиации;5. беременным женщинам с синдромом Марфана необходимо один раз в 2 мес проводить эхокардиографию. При диаметре аорты 45 мм и выше следует безотлагательно решать вопрос о целесообразности дальнейшего сохранения беременности;6.
родоразрешение женщин с синдромом Марфана необходимо осуществлять с помощью кесарева сечения в специализированных родильных домах для рожениц с патологией сердечно-сосудистой системы.
Профилактика синдрома Марфана
Больным с синдромом Марфана, вступающим в брак, показано медико-генетическое консультирование, информация о степени повторного риска развития у детей аналогичного заболевания. Наряду с этим, необходима также пре-натальная диагностика.
Врач терапевт Жумагазиев Е.Н.
Источник: https://medicalj.ru/diseases/congenital-anomaly/918-sindrom-marfana

