
Синдром Марфана (продолжение…)
Синдром Марфана, или человек-паук: случай из практики
Синдром Марфана заболевание исключительно наследственного характера и передаётся от родителей, которые имеют в своём арсенале здоровья данное заболевание. При данном синдроме происходит нарушение развития соединительной ткани из-за мутаций генов, которые кодируют образование гликопротеина фибриллина-1.
Частота возникновения и проявления заболевания различается.
В статье:
Доподлинно известно, что синдромом Марфана болели сказочник Ганс Христиан Андерсен, композитор и музыкант Никколо Паганини (написанные им произведения может исполнять не каждый пианист или скрипач, поскольку порой не хватает разведения пальцев руки для того, чтобы взять тот или иной аккорд).
История появления
Впервые болезнь описана и впоследствии названа Антуаном Марфаном — французским профессором педиатрии, который наблюдал маленькую девочку с необычными аномалиями костей скелета, причём данные аномалии с каждым годом усугублялись, то есть болезнь прогрессировала.
Признаки синдрома Марфана
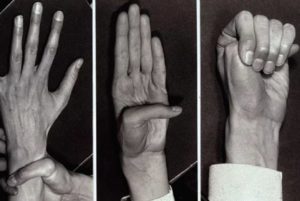
- У людей с синдромом Марфана обычно непропорционально длинные конечности, так называемая долихостеномелия.
Помимо этих особенностей нередко встречаются деформации позвоночника и грудной клетки в виде вдавленной или куриной груди, слабость мышц, плоскостопие и высокое нёбо;
- Имеется гипермобильность суставов, то есть суставы патологически подвижны;
- Кроме того бросаются в глаза очень длинные пальцы, будто паучьи лапки – арахнодактилия (вспомните о невозможности взять тот или иной аккорд при исполнении произведений Н. Паганини – именно из-за длинных его пальцев);
- Также недоразвит подкожно-жировой слой;
- Нарушения зрения в виде подвывиха хрусталика и отслойки сетчатки;
- Нарушения работы сердечно-сосудистой системы: пролапс митрального клапана встречается у 80%, аневризма аорты;
- В области плеч, поясницы и груди часто возникают стрии;
- В почках и печени кисты и кистозные разрастания.
Нужно отметить, что все выше приведенные признаки заболевания могут проявляться в разных степенях тяжести, причём в одних случаях эти изменения практически незаметны, а в других приводят к угрожающим жизни состояниям.
Частота встречаемости и прогноз заболевания
Встречается синдром не очень редко – в одном случае на 5000 человек.
При отсутствии лечения люди с синдромом Марфана могут дожить максимально до сорока лет. Смерть наступает в основном из-за расслаивающей аневризмы аорты. При эффективном и адекватном лечении пациенты доживают до пожилого возраста.
Лечение синдрома Марфана
Этиологического лечения, то есть лечения причины возникновения данного заболевания на данный момент нет. В основном на этом этапе развития медицины осуществляется симптоматическое лечение и профилактика развития острых состояний, вызванных декомпенсацией сердечно-сосудистой системы и в меньшей степени других систем.
Это кратко о синдроме Марфана. Теперь позвольте поделиться опытом работы с пациентом, который страдает данным заболеванием.
Клинический случай
На данный момент у нас уже очень длительное время находится один пациент с синдромом Марфана, однако это заболевание у него не единственное.
Молодому человеку уже чуть больше двадцати лет и он никогда не мог ни ходить, ни поворачиваться в кровати, ни говорить, ни самостоятельно есть. К нему никогда не приходили родственники, но родители где-то есть. У него имеются большие проблемы с психической и физической сферой, которые проявляются тяжёлыми проблемами с интеллектом и опорно-двигательным аппаратом.
Как контактирует с окружающим миром?
Конечно, контакт с людьми затруднён, поскольку он не может выразить своих эмоций, не может ничего сказать или рассказать.
Понять его может только человек, который уже длительное время за ним ухаживает, поскольку у пациента есть свои чёткие особенности поведения.
К примеру, он любит играть с пелёнкой и некоторыми определёнными игрушками. Когда он хочет играть и когда не хочет тоже нужно иметь опыт рассмотреть.
Как он ест?
Кушает молодой человек только протёртую пищу. Кормят его из ложки очень аккуратно, потому что он достаточно часто попёрхивается, что опасно для жизни. У него есть свои пристрастия в еде, но практически всегда он ест хорошо и с аппетитом.
Молодой человек обычно кормится в полусидячем положении, поскольку кормить в горизонтальном таких пациентов нельзя.
Несмотря на достаточное количество принимаемой пищи у него имеется недостаток массы тела.
Усваиваемость питательных веществ находится не на самом высшем уровне и периодически в крови у него падает общий белок и альбумин, что приводит к отёкам нижних конечностей.Данные состояния корректируются назначением дополнительного высокоэнергетического энтерального питания, которое у нас имеется в крайне ограниченном количестве.
Как часто болеет?
Он почти никогда не болеет инфекционными заболеваниями и простудами, несмотря на проблемы с нутритивным статусом. В основном из болезней у него анемия и недостаток массы тела. Анемия у него проявляется достаточно часто и всегда лечится препаратами железа.
Стул у него регулярный и достаточный благодаря слабительным свечам, а количество мочи всегда в норме само по себе.
Трудности в уходе
Достаточно сложно избегать пролежней. Необходимо постоянно переворачивать его с одного бока на другой, что делать крайне затруднительно из-за его контрактур.
Также ухаживающий взрослый человек испытывает трудности с кормлением особенного пациента.
О реабилитационном потенциале
Конечно, на сегодняшний день реабилитационный потенциал отсутствует и не факт, что он когда-либо был.
Стоит отметить, если бы у нашего пациента с синдромом Марфана были хорошие активные родители и занимались бы с ним с самого рождения, возможно, какие-то положительные моменты в физической реабилитации и социальной адаптации появились бы и подняли этого человека на более высокий уровень. Однако это всего лишь если и как оно было бы на самом деле неизвестно.
На сегодняшний день как таковой реабилитации быть не может, но могут быть занятия направленные на внесение в жизнь человека разнообразия, дабы ему не было тоскливо и скучно.
За годы работы с такими тяжёлыми детьми и людьми пришло чёткое осознание того, что они прекрасно всё понимают, просто вторая сигнальная система (речь ) неразвита и они не могут нам ничего рассказать о своём состоянии.
В заключение
Мы вкратце представили вашему вниманию уникальный практический случай, как он есть на самом деле. Если у вас есть вопросы, если вас волнуют проблемы, затронутые в статье, пишите нам и мы вам обязательно ответим. Спасибо за внимание.
Источник: http://www.childrenspace.by/meditsina/redkie-sindromy/item/126-sindrom-marfana-ili-chelovek-pauk-sluchaj-iz-praktiki.html
Болезнь Марфана
Болезнь Марфана – наследственное заболевание, характеризующееся поражением соединительной ткани. Эту патологию еще называют синдромом Марфана. Поражается не только соединительная ткань, но и скелетно-мышечная система, зрение и многое другое.
Соединительная ткань необходима для соединения определенных частей органов. Благодаря ей закладываются основы для нормального функционирования органов и систем организма, их естественного развития. При возникновении болезни Марфана в структуре соединительной ткани появляются такие дефекты, которые препятствуют ее нормальному развитию и функционированию.
В последние годы заболевания, имеющие наследственный фактор, становятся более частыми. Есть гипотеза, что так происходит из-за накопления человеком изменений на уровне ДНК.
Этому могут способствовать плохая экология, аномальный климат, некачественные продукты питания и повсеместное облучение сотовой и мобильной связью.
Однако такая гипотеза не доказана, а количество патологических наследственных заболеваний продолжает увеличиваться.
Так как соединительная ткань расположена по всему телу, то пациенты с синдромом Марфана могут иметь в анамнезе такие заболевания:
- аневризму аорты или легочной артерии;
- гигантизм;
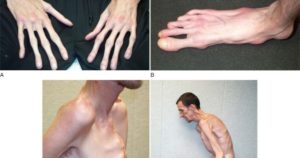
- арахнодактилию или долихостеномелию;
- протрузию позвонков или вертлужной впадины;
- патологию твердой мозговой оболочки;
- нарушение зрения (например, эктопия хрусталика);
- сколиоз позвоночного столба;
- поражения сердечно-сосудистой и центральной нервной системы;
- деформацию грудной клетки и многие другие.
Болезнь Марфана отличается вариабельностью симптоматики. Иногда рождаются люди с такими патологиями, которые несовместимы с жизнью. В таких случаях пациенты гибнут сразу при рождении или через несколько лет жизни. Есть те, кто имеет скрытые формы синдрома Марфана.
Заболевание это довольно редкое. По статистике в Европейских государствах встречается в 2-х случаях на 10000 человек.
Причины возникновения и предрасполагающие факторы
причина болезни Марфана – изменения в гене (15-я хромосома), который отвечает за выработку белка фибриллина в соединительной ткани. Данный белок важен в организме тем, что придает соединительной ткани растяжимость, эластичность, упругость.
Если нормальное строение нарушается, изменяется, это приводит к тому, что ткань не имеет достаточной упругости и прочности. Если данный фактор резко выражен, человек не способен переносить даже самые маленькие физически нагрузки, потому что ткань не выдерживает их.
Особенно негативно нарушения соединительной ткани сказываются на состоянии связочного аппарата, стенок сосудов, тканей.
Предрасполагающим фактором может быть наследственность. Также возможен спорадический случай рождения такого больного, когда в поколении родных пациента такого видоизменения гена еще не было.
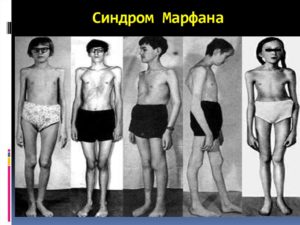
Пациенты с данным синдромом почти всегда очень высокие. Имеют тонкие длинные конечности рук и ног, а само туловище непропорционально короткое. При этом глаза у них впалые, наблюдается неправильное образование зубных рядов (нижняя челюсть иногда сильно выступающая), уши также большие, пальцы длинные и тонкие.
Возможные последствия
Деформация в любом случае затрагивает многие части тела и внутренние органы. Это негативно сказывается на здоровье, общем самочувствии человека и его внешнем виде.
Последствия самые различные:
- воронкообразная грудная клетка (или килевидная, происходит за счет развития деформации в грудной клетке);
- развитие любых форм сколиоза (кифоз, кифосколиоз, др.);
- спондилолистез;
- патологии костно-мышечной системы: разболтанность многих суставов, протрузии, слабость мышечной ткани;
- пороки развития сердца;
- дилятация стенок аорты и расслоение;
- стеноз легочной аорты;
- нарушение сердечного ритма;
- частая отслойка сетчатки.
Это далеко не полный список возможных заболеваний и патологий, развивающихся вследствие болезни Марфана. Ввиду серьезности поражений внутренних органов и сердца, такие пациенты редко живут долго. Они имеют отклонения и в психомоторном развитии, причем уровень интеллекта может быть очень низким, а может быть достаточно высоким, но это редкий факт.
Часто наблюдаются инсульты, инфаркты головного мозга и/или сердца, частые обмороки, сильные головокружения. Связано это с нарушением кровообращения и патологией сосудистой системы.
Важно! Пациенты с синдромом Марфана должны постоянно проходить диагностику и профилактическое лечение. От этого зависит качество и продолжительность их жизни.
Классификация болезни Марфана
- По характеру заболевание бывает прогрессирующим и легким.
- По тяжести симптомов и протекания: легкая, средняя и тяжелая степени тяжести.
- По форме – скрытая (стертая) и выраженная.
Диагностика болезни Марфана
Заболевание диагностируется при клиническом осмотре, на основе лабораторных анализов и инструментальных исследований. Немаловажное значение имеет семейный анамнез.
Для уточнения диагноза проводятся:
- эхокардиография (для обнаружения клапанной регургитации, измерения размеров левого желудочка, разрывы хорд); электрокардиограмма;
- аортография (при подозрении на расслоение аорты);
- рентген грудной клетки, легких, позвоночного столба (наличие протрузий, кист, измерение дуги аорты и размеров сердечной мышцы);
- МРТ и/или КТ отделов головного мозга и сердца (проверяется возможное наличие дилатации и аневризмы аорты, новообразования в отделах головного мозга, др.);
- анализы крови и мочи.
Зачастую для определения диагноза много исследований не нужно, его можно поставить сразу на основе физикального осмотра. В других случаях необходимы консультации многих узконаправленных специалистов: ортопеда или артролога, кардиолога, окулиста, невролога, психотерапевта, генетика.
Лечение и профилактика
Исправить консервативным лечением наличие серьезных врожденных патологий невозможно. Иногда назначается хирургическое лечение для исправления той или иной ситуации, например, сильной разболтанности коленного сустава. Если в целом здоровье пациента нормальное, проводится артроскопия.
Также, если в целом здоровье позволяет, больным выполняют и другие оперативные вмешательства, например, на сердце (при пороке сердца, нарушении в работе сердечных клапанов, протезирование митрального клапана) и глазах (при глаукоме, катаракте, астигматизме, вплоть до имплантации искусственного хрусталика). Применяется коллагеннонормализующее восстановление, метаболическая терапия, рекомендуется прием витаминов и минералов.
Основное лечение направлено на улучшение состояния пострадавших органов, укрепление работы сердца, нормализацию кровообращения.Проводится профилактическое лечение для предупреждения возникновения других болезней. Таким больным показан частый врачебный осмотр и исследования всего организма, чтобы своевременно устранить возникновение серьезных причин для заболеваний, предупредить возникновение новых.
Прогноз жизни при болезни Марфана
Средняя продолжительность жизни людей с данным синдромом – примерно 40-45 лет. Многое зависит от тяжести поражения сердечно-сосудистой системы. Также важно состояние скелета и мышц.
Риск осложнений всегда высокий, потому и внезапная смерть у многих достаточно частая. Некоторые не доживают и до 30 лет. Есть случаи, когда больные синдромом Марфана жили более 69 лет, однако такие пациенты всегда проходили тщательную диагностику и современное лечение.
Галина Владимировна
Источник: http://comp-doctor.ru/zoj/marfan.php
Синдром Марфана
- Определение
- Причины
- Симптомы
- Диагностика
- Профилактика
Синдром Марфана — аутосомно-доминантное заболевания соединительной ткани, при котором наблюдают различные патологические изменения сердечно-сосудистой системы, кожи, скелета, органов зрения, легких и твердой мозговой оболочки.
Причины
Частота заболевания составляет порядка 2-3 на 10 тыс., из которых 25-30% — это новые мутации.
Синдром Марфана обусловлено мутацией гена N1 в хромосоме 15q21, что приводит к дефициту фибриллина-1, гликопротеина во внеклеточной матрице и нарушения регуляции трансформированного фактора роста ß (ТФР-(ß).
Известно более 1000 мутаций, почти все они уникальны для пораженной семьи. У 10% пациентов с точным диагнозом синдрома Марфана мутацию N1 все еще не определено.
Симптомы
Прогноз обычно определяется расширением аорты, которое приводит к диссекции или ее разрыву, которые являются основными причинами смерти. Средняя продолжительность жизни пациентов без лечения составляет 40 лет. Расширение корня аорты обнаруживают в 60 — 80% пациентов.
Риск диссекции аорты типа А явно повышается с расширением диаметра корня аорты, но диссекции может иногда возникать даже у пациентов с небольшой дилатацией. Пациенты с расширенной аортой обычно бессимптомные.
Наличие значительного регургитации на аортальном, трикуспидальном или митральном клапане может привести к появлению объемной перегрузки ЛЖ, но возможно его независимое поражения.
Синдром Марфана требует дифференциальной диагностики с другими наследственными болезнями соединительной ткани, симптомы которых очень похожи к симптомам синдрома Марфана, такими как синдром Лойса — Дитца, семейная аневризма аорты, двустворчатый аортальный клапан с расширением аорты, семейная эктопия хрусталика, МSS фенотип и синдром Элерса Данлоса.
Диагностика
Раннее установление диагноза позволяет предотвратить диссекции аорты и ее разрыва, но требует работы многопрофильной команды врачей.
Сейчас диагноз синдрома Марфана устанавливают при наличии значительных поражений органов двух различных систем, важнейшие из которых — аневризма/диссекции корня аорты и эктопия хрусталика с привлечением органов третьей системы (нозология Гента), критерии которой недавно пересмотрены. На сегодня большее значение имеет молекулярно-генетическое тестирование.
С помощью эхокардиографического исследования необходимо определять максимальный диаметр аорты, ее размер на уровне фиброзного кольца, синусов Вальсальвы, синотубулярного соединения и дальнейшей восходящей части аорты, оценивать функцию ЛЖ, аортального, митрального и трикуспидального клапанов. Эхокардиография необходима в случае подозрения на диссекцию аорты или перед вмешательством.
МРТ или КТ аорты нужно проводить каждому пациенту. Показатели эластичности нисходящей части аорты, по данным МРТ, является независимым предиктором прогрессирования дилатации нисходящей части аорты.Поскольку эндоваскулярные манипуляции создают риск перфорации стенки аорты, коронарная ангиография для выявления ИБС желательно производить непосредственно перед операцией.
Симптомно пациенты требуют применения холтеровского мониторирования для своевременной диагностики желудочковых аритмий, нарушений проводимости и предотвращения внезапной сердечной смерти (ВСС).
Профилактика
Пациенты нуждаются пожизненного регулярного наблюдения в специализированных центрах. Эхокардиографическое исследование с определением функции клапанов и желудочков у стабильных пациентов должны проводить ежегодно.
Визуализация аорты с помощью МРТ (или КТ, если МРТ противопоказана) в начале наблюдения и раз в 5 лет, если размер аорты за ее корнем нормальный, и ежегодно, если есть аневризма выше корня аорты, чрезвычайно важна, особенно, если осталась диссекции.
Пациентам следует рекомендовать воздерживаться от физических упражнений значительной интенсивности, спортивных соревнований, контактных видов спорта и изотермических нагрузок.
Вероятность наследования синдрома Марфана от матери составляет 50%, что обусловливает потребность в генетической консультации.
Женщин с диаметром аорты> 45 мм следует отказываться от беременности до коррекции порока, поскольку риск диссекции высок. По размеру аорты от 40 до 45 мм.
для определения целесообразности хирургического вмешательства до беременности нужна информация о динамике предварительной дилатации аорты и семейный анамнез. Однако опасность существует при любом диаметра аорты.
Профилактика ИЭ рекомендована только пациентам с высоким риском.
Медикаментозное лечение
Современное медикаментозное и хирургическое лечение продлило продолжительность жизни больных с синдромом Марфана до 60 — 70 лет. Бета-адреноблокаторы могут уменьшить риск дилатации аорты и улучшить выживаемость, по крайней мере у взрослых. Важно тщательное медикаментозное лечение АГ с целевым уровнем систолического АД
Источник: http://med36.com/ill/1161

