
Положительная проба Фелинга у ребенка 2.5 года.
Фенилкетонурия

Заболевание передаётся по аутосомно-рецессивному типу. То есть ребёнок должен наследовать две копии мутантного гена: одну – от матери, вторую – от отца. При наследовании только одной копии ребёнок, как и его родители, будет носителем болезни.
Если оба родителя являются носителями фенилкетонурии, то шансы родиться больными у их детей равны один к четырём – ровно столько же, как и возможность родиться абсолютно здоровым. Шанс родиться носителем болезни у детей таких родителей составляет 2/4. Фенилаланингидроксилаза – очень важный печёночный фермент, который преобразует фенилаланин в тирозин.
Вследствие мутации гена, который в норме должен контролировать синтез фенилаланингидроксилазы, образуется блок синтеза этого фермента или он синтезируется в критически малых количествах.
Это приводит к тому, что фенилаланин начинает дезаминироваться (происходит процесс отщепления аминогрупп от молекулы органического соединения) и синтезировать очень токсичные вещества в организме: фенилуксусную, фенил-молочную и фенилпировиноградную кислоты.
Кроме того, недостаток тирозина приводит к нарушению образования меланина и катехоламинов – веществ, которые выполняют роль «управляющих» молекул в области межклеточных взаимодействий во всём организме и особенно в клетках головного мозга.
Очень редко (1% от всех случаев заболевания) фенилкетонурия развивается из-за мутации в генах, которые контролируют синтез кофактора фенилаланингидроксилазы — BH4 (тетрагидробиоптерина). В результате данной мутации в организме отсутствует или присутствует в очень малых количествах BH4, что приводит к невозможности выполнения своих функций фенилаланингидроксилазой и развитии всех вышеперечисленных вытекающих явлений. Данный тип фенилкетонурии также передаётся по аутосомно-рецессивному типу.
Симптомы фенилкетонурии
Симптомы возникают уже на первых неделях, реже – месяцах жизни ребёнка и проявляются в разных видах. — Чрезмерная сонливость. — Вялость, апатия. — Вместо сонливости может быть, наоборот, плаксивость и капризность. — Отмечается значительная отсталость в психическом и физическом развитии.
— В дальнейшем могут развиваться эпилептиформные припадки любого характера (судорожные, отключение сознания или бессудорожные движения конечностей или головы, вздрагивания). — В процессе развития болезни возникает тонус отдельных определённых групп мышц, из-за чего ребёнок приобретает вынужденную позу «портного» – согнутые руки и поджатые ноги.
— В тяжёлых случаях могут развиваться тремор рук, гиперкинезы (непроизвольные, внезапно возникающие, резкие, хаотические движения), атаксия (нарушение координации движений). — Обычно больные дети белокурые, с голубыми глазами и белой чувствительной кожей, склонной к экземам и дерматитам. — Часто отмечается у детей сильная потливость с характерным «мышиным запахом».
— В процессе развития болезни моча приобретает «мышиный запах». — Если не начать лечение, то развиваются имбецильность, или идиотия.
Диагностика фенилкетонурии
Для обнаружения заболевания до момента появления первых симптомов всем новорожденным проводят скрининг по специальным профаммам, позволяющий выявить повышенный уровень фенилаланина в крови ещё на первых неделях жизни ребёнка.
Если у ребёнка обнаруживаются самые минимальные признаки возможного наличия фенилкетонурии, необходимо немедленно сдать анализ на определение уровня фенилаланина в крови и/или сделать пробу Фелинга.
Проба Фелинга
К моче ребёнка (можно провести тест прямо на пелёнке, запачканной мочой малыша) добавляют уксусную кислоту и 5%-ный раствор треххлористого железа. Моча приобретает зелёный цвет. Эти методы исследования считаются ориентировочными. При положительном результате необходимо дополнительное обследование, направленное на точное определение количества фенилаланина в моче и крови, если ребёнок старше двух недель жизни, или провести определение активности фенилаланингидроксилазы в биоптате (материале, взятом при помощи биопсии) печени и/или анализ ДНК на выявление мутантного гена, если ребёнок младше или старше двух недель. При подозрении на фенилкетонурию, связанную с мутацией гена, регулирующего синтез кофактора BH4, необходимы дополнительные исследования. В частности, необходим анализ ДНК, нацеленный на поиск мутантного гена.
Лечение фенилкетонурии
Направлено на снижение содержания фенилаланина в крови. Для этого ребёнку назначается специальная диета, исключающая продукты, содержащие фенилаланин, то есть вся пища животного происхождения.
Дефицит белка восполняется белковыми гидролизатами (аминокислотными смесями) – лефанолаком, цимограном, гипофенатом, берлофеном. До полового созревания (до возраста не менее 18 лет) эти смеси становятся основным продуктом питания для ребёнка. Их можно вводить вместе с супами, фруктовыми соками, овощными пюре.
Некоторые фрукты и овощи, содержащие фенилаланин, тоже исключаются из рациона питания. На протяжении всего лечения необходим строгий контроль уровня фенилаланина в крови. Многие учёные считают, что диета необходима больным на протяжении всей жизни.Если болезнь определить в доклинической стадии и вовремя назначить лечение, то развития симптомов можно полностью избежать. При отсрочке терапии наступают необратимые повреждения тканей мозга.
Профилактика фенилкетонурии
Выявление носительства мутантного гена у обоих родителей и ЭКО (экстракорпоральное оплодотворение), а также скрининг всех новорожденных для своевременного назначения лечения и недопущения развития болезни.
Источник: http://www.happy-giraffe.ru/childrenDiseases/phenylketonuria/
Фенилкетонурия | Диагностика и лечение фенилкетонурии
Известно достаточное количество нарушений обмена веществ, в том числе отдельных аминокислот. Большинстводанных нарушений обмена имеют наследственный или врожденный характер.Таким примером являетсяфенилкетонурия.
Фенилкетонурия (фенилпировиноградная олигофрения) – это заболевание с аутосомно-рецессивным типом наследования из группы ферментопатий, которое развивается в результате потери способности организма синтезировать фермент фенилаланин-4-монооксигеназу, способствующегопревращениюаминокислоты фенилаланина в тирозин, и приводит к тяжелому поражению центральной нервной системы (ЦНС) с нарушением умственного развития ребенка.
Средняя частота данной патологии у новорожденных составляет около 1:10 000; при этом мальчики и девочки одинаково частоболеют, но мужской пол чаще погибает на 1-м году жизни.
Выделяются:
- Типичная форма, или ФенилкетонурияI(недостаток фермента фенилаланин-4-монооксигеназы);
- Атипичные формы:
- ФенилкетонурияII (дефицитдигидроптеридинредуктазы);
- ФенилкетонурияIII(недостаток 6-пирувоилтетрагидроптерин синтетазы).
Причины фенилкетонурии
Причина фенилпировинограднойолигофрении – аутосомно-рецессивно унаследованный недостаток фермента фенилаланин-4-монооксигеназы (-гидроксилазы), синтезируемого в печени, в результате чего метаболическое превращение аминокислоты фенилаланина в тирозин не происходит.
Результат этого блокирования – накопление фенилаланина и его кетопроизводных(фенилмолочной, фенилуксусной и фенилпировиноградной кислот) в организме с появлением их в очень большом количестве. В норме данные вещества не образуются. Кроме того, в головном мозге образуются ортофенилацетат и фенилэтиламин, избыток которых способствует нарушению липидного обмена в головном мозге.
Все это ведет к прогрессирующему снижению интеллекта вплоть до идиотии у данных больных.
Обнаружены также дефекты обмена биоптерина – недостаток 6-пирувоилтетрагидроптерин синтетазы, которые приводят к развитию атипичных форм фенилпировинограднойолигофрении. Данные нарушения составляют не более 10% случаев фенилкетонурии.
Важнейшие особенности заболевания:
- замедление умственного развития ребенка;
- выделение с мочой огромных количеств фенилпирувата(до 1–2 г/сут) и фенилацетилглутамата (до 2–3 г/сут).
Признаки заболевания появляются уже на первой неделе или месяце жизни ребенка.
Дети резко отстают в умственном и нервно-психическом развитии. Отмечается сонливость, вялость, плаксивость, повышенная раздражительность. Если болезнь прогрессирует, то возможно развитие эпилептиформных припадков – поклоны, кивки, вздрагивания развернутого судорожного или бессудорожного типа.
Возможно появление кратковременного отключения сознания.
Для ребенка характерна опреденная поза – поджатые ноги и согнутые руки («поза портного») из-за гипертонусаопреденных групп мышц. Могут появляться гиперкинезы, атаксия, тремор рук, парезы центрального типа.
Нередко дети имеют характерный внешний вид – светлокожие с голубыми глазами. У них часто отмечается развитие кожных заболеваний, таких как экзема, дерматиты. Кожа склонна к чрезмерной травматизации. Появляется своеобразный мышиный запах мочи и пота.
Обнаруживается склонной к пониженному артериальному давлению (гипотензии). Нередко у детей обнаруживаются пороки сердца, гипофизарный нанизм, гипогонадизм, уменьшение размеров черепа, расстройства координации движений, вегетативные дисфункции, нарушения походки, поведения.
При отсутствии адекватного лечения развивается глубокая психическая инвалидность, имбецильность, идиотия.
Диагностика фенилкетонурии в Израиле
- Микробиологический тест Гатрии флюорометрический методы (важнейшее доказательство фенилкетонурии – данные о накоплении фенилаланина в тканях):
- в крови количество фенилаланина может достигать 600 мг/л (когда в норме 15 мг/л);
- в цереброспинальной (спинальной) жидкости– 80 мг/л (когда норма 1,5 мг/л).
- Характерная особенность болезни – это экскреция (выделение) с мочой огромных количеств фенилпирувата (до 1–2 г/сут) и фенилацетилглутамата (до 2–3 г/сут). Но этот тест проводится только с 10-12 дня после рождения ребенка.
- Проба Фелинга – обнаружениефенилкетонурии с помощью 5-10 % раствора хлорида железа (FeCl3) и уксусной кислоты: появление оливково-зеленойокраски мочи спустя 2–3 мин после добавления в нее нескольких капель 5-10 % раствора FeCl3и уксуской кислоты. Также применяются индикаторные бумажки «Биофан», «Фенистикс».
- Биопсия печени (взятие участка ткани печени) – исследование ткани печени на активность фермента фенилаланин-4-монооксигеназы.
- Хроматография аминокислот.
- Использование аминоанализаторов (количественнаяконцентрацияфенилаланина в крови).
Лечение фенилкетонурии в Израиле
Значительно снизив прием с пищей с самого рождения ребенка фенилаланина можно предотвратить развитие болезни.
Поздно начатое лечение часто даёт определённый эффект, но никогда не устраняет ранее развившихся измененийткани мозга, которые необратимы.
Лечение включает в себя:
- Строгое соблюдение диеты (от обнаружения болезни и как минимум до полового созревания ребенка): исключаются все молочные, мясные и рыбные продукты, которые содержат животный и , частично, растительный белок.
- Восполнение белка аминокислотными смесями, в которых отсутствует фенилаланин (белковые гидролизаты:Лефанолак, Берлофен, Гипофенат).
- Хороший эффект дает лечение атипичных форм фенилкетонурии фактором пораженного фермента – тетрагидробиоптерином.
- Применяются новые методы лечения фенилпировиноградной олигофрении:
- заместительная терапия фенилаланинлиазой (PAL) – ферментом растительного происхождения, который превращает фенилаланин в безвредные продукты обмена;
- генотерапия- введение в организм больного вирусного вектора, который содержит ген фенилаланин-4-гидроксилазы.
Внимание все поля формы обязательны. Иначе мы не получим вашу информацию. Альтернативно пользуйтесь info@hospital-israel.ru
Источник: https://hospital-israel.ru/nevrologia/bolezni/fenilketonuriya/
Фенилкетонурия
Фенилкетонурия – наиболее распространенное нарушение обмена аминокислот. В среднем фенилкетонурии подвержен 1 из 8000 человек.
В основе болезни лежит дефицит фермента, осуществляющего превращение фенилаланина в тирозин (тирозин препятствует отложению жиров, снижает уровень аппетита, улучшает функции гипофиза, щитовидной железы и надпочечников).
Симптомы фенилкетонурии
Фенилкетонурия проявляется на первом году жизни. Основными симптомами в этом возрасте являются:
- вялость ребенка;
- отсутствие интереса к окружающему;
- иногда повышенная раздражительность;
- беспокойство;
- срыгивания;
- нарушения мышечного тонуса (чаще мышечная гипотония);
- судороги;
- признаки аллергического дерматита;
- появляется характерный «мышиный» запах мочи.
В более позднем возрасте для больных фенилкетонурией характерна задержка психоречевого развития, нередко отмечается микроцефалия.
При фенилкетонурии характерны следующие фенотипические особенности: гипопигментация кожи, волос, радужной оболочки глаз. У некоторых больных одним из проявлений патологии может быть склеродермия.
Эпилептические приступы встречаются почти у половины больных фенилкетонурией и в некоторых случаях могут служить первым признаком болезни.
Диагностика
Диагноз при подозрении на фенилкетонурию основывается на совокупности генеалогических данных, результатов клинического и биохимического обследования:
- возможный родственный брак родителей больного ребенка;
- аналогичная патология у родных или двоюродных сибсов (братьев или сестер);
- судороги, нарушение мышечного тонуса;
- экзематозные изменения кожи;
- гипопигментация волос, кожи, радужной оболочки глаз;
- своеобразный «мышиный» запах мочи;
- повышенный уровень фенилаланина в крови > 900 мкмоль/л;
- присутствие в моче фенилпировиноградной, фенилмолочной, фенилуксусной кислот;
- положительная проба Феллинга.
В настоящее время для диагностики фенилкетонурии разработаны и внедрены молекулярно-генетические методы выявления генного дефекта.
Диагностика у новорожденных (скрининг)
В связи с достаточной распространенностью фенилкетонурии, тяжестью клинических проявлений и реальной возможностью профилактического лечения, фенилкетонурия в числе первых наследственных нарушений обмена веществ была включена в список наследственных заболеваний, рекомендованных Всемирной организацией здравоохранения для раннего выявления среди новорожденных.
Для ранней диагностики фенилкетонурии в России осуществляется массовое обследование детей в родильных домах с определением уровня фенилаланина в крови.
Забор крови производится у новорожденных в возрасте 4–5 дней. Реже объектом исследования является моча.
Лечение фенилкетонурии
Главным способом лечения фенилкетонурии является диетотерапия, ограничивающая поступление в организм белка и фенилаланина.
Основным критерием адекватности диеты при фенилкетонурии служит уровень фенилаланина в крови, который должен:
- в раннем возрасте составлять 120–240 мкмоль/л;
- у детей дошкольного возраста – не превышать 360 мкмоль/л;
- у школьников – не превышать 480 мкмоль/л;
- у детей старшего школьного возраста допустимо увеличение содержания фенилаланина в крови до 600 мкмоль/л.
Пищевой рацион строится путем резкого ограничения поступления белковых продуктов животного и растительного происхождения и, следовательно, фенилаланина. Для облегчения расчетов принято считать, что 1 г условного белка содержит 50 мг фенилаланина.
При лечении фенилкетонурии полностью исключают продукты, богатые белком и фенилаланином: мясо, рыбу, сыр, творог, яйца, бобовые и др. В пищевой рацион больных входят овощи, фрукты, соки, а также специальные малобелковые продукты – амилофены.
Для коррекции белкового питания и восполнения недостатка аминокислот при фенилкетонурии назначаются специальные лечебные продукты:
- белковые гидролизаты: нофелан (Польша), апонти (США), лофенолак (США);
- смеси L-аминокислот, лишенные фенилаланина, но содержащие все другие незаменимые аминокислоты: фенил-фри (США), тетрафен (Россия), П-АМ универсальный (Великобритания).
Несмотря на обогащение аминокислотных смесей и белковых гидролизатов минеральными и другими веществами, больные фенилкетонурией нуждаются в дополнительном назначении витаминов, в частности группы В, минеральных соединений, особенно содержащих кальций и фосфор, препаратов железа и микроэлементов.
В последние годы для страдающих фенилкетонурией была обоснована необходимость применения препаратов карнитина (L-карнитин, элькар в средней суточной дозе 10–20 мг/кг массы в течение 1–2 мес. 3–4 курса в год) для профилактики его недостаточности.
Параллельно лечение фенилкетонурии осуществляется медикаментозным патогенетическим и симптоматическим лечением ноотропными средствами, препаратами, улучшающими сосудистую микроциркуляцию, по показаниям – антиконвульсантами.
Широко используется лечебная гимнастика, общий массаж и др. Комплексная реабилитация детей с фенилкетонурией предусматривает специальные методы педагогических воздействий в процессе подготовки к школе и школьного обучения. Больные нуждаются в помощи логопеда, педагога, в ряде случаев – дефектолога.Большие споры вызывает вопрос о длительности диетотерапии в лечении фенилкетонурии.
В последнее время большинство врачей принимает точку зрения о необходимости продолжительного выполнения диетических рекомендаций.
Обследование детей, прекративших соблюдать диету в школьном возрасте, и детей, продолжавших получать диетотерапию, однозначно показало значительно более высокий уровень интеллектуального развития последних.
У больных фенилкетонурией старшего возраста, в том числе подростков, безусловно, возможно постепенное расширение диеты в связи с улучшением толерантности к фенилаланину.
Коррекция питания осуществляется, как правило, путем введения в рацион ограниченного количества круп, молока и некоторых других натуральных продуктов, содержащих относительно умеренное количество фенилаланина.
В период расширения рациона проводятся оценка нервно-психического статуса детей, контроль электроэнцефалограммы, уровня фенилаланина в крови.
В возрасте старше 18–20 лет проводится дальнейшее расширение диеты, однако и во взрослом периоде пациентам рекомендуется отказаться от высокобелковых продуктов животного происхождения.
Особенно строго подходят к диетотерапии девочек, страдающих фенилкетонурией, и женщин в репродуктивном периоде. Такого рода больным фенилкетонурией необходимо продолжать диетическое лечение для обеспечения рождения здорового потомства.В последние годы разрабатывается способ снижения уровня фенилаланина в крови путем приема препарата, содержащего фенилаланингидроксилазу растительного происхождения.
Данная статья опирается на статью из книги «Врожденные и наследственные заболевания» под редакцией профессора П.В.Новикова, М., 2007
Источник: https://www.diagnos.ru/diseases/gena/fenilketonuria
Фенилкетонурия у детей: причины возникновения и симптомы у новорожденных
ФКУ (аббревиатура заболевания фенилкотурия) — это редкий наследственный недуг, причина возникновения которого кроется в расстройстве обмена аминокислот. Организм не в состоянии утилизировать фенилаланин. Он поступает вместе с пищей и накапливается в теле пациента и медленно отравляет его изнутри.
Подобно яду, он действует на нервную систему больного и вызывает соответствующую симптоматику. Вследствие этого, ФКУ имеет и другое название – фенилпировиноградная олигофрения. Это, пожалуй, единственное наследственное заболевание, которое можно целиком купировать. Дети, рожденные с этим диагнозом, при должном лечении вырастают нормальными и здоровыми.
Фенилкетонурия у новорожденных достаточно редко встречается. В разных странах этот показатель отличается. Так, в России 1 случай заболевания приходится на 10 000. Великобритания имеет большую частоту данного показателя – 1 на 5 000. В Африке патология практически не встречается. Согласно статистике, девочки болеют ФКУ чаще, чем мальчики примерно в 2 раза.
Причины возникновения
Пусковым фактором для развития заболевания является наследственность. ФКУ зарождается в двух случаях:
- При близкородственных браках повышается вероятность проявления дефектных генов, в том числе растет шанс возникновения фенилкетонурии;
- Из-за мутации в области 12 хромосомы ввиду разнообразных причин.
ФКУ возникает спонтанно в, казалось бы, благополучной семье, без известных случаев данного заболевания у родственников. Наличие патологического гена в организме пациента приводит к тому, что печень не вырабатывает специальный фермент фенилаланин – 4 – гидроксилазу. Это и является причиной развития клинических проявлений заболевания.
Патогенез
Фенилкетонурия возникает в тех случаях, когда оба родителя – папа и мама, передали малышу склонность к этой патологии. Они сами могут не болеть, но сочетание у ребенка двух дефектных генов от родителей наделяют его шансом к ФКУ 25%. Вероятность такого совпадения довольно мала, поэтому и феникетонурия встречается нечасто.
Фермент, отсутствующий при болезни, должен превращать поступающую с белковой пищей аминокислоту фенилаланин в тирозин.
Последний составляет пигмент меланин, другие ферменты, гормоны, то есть нужен для адекватной работы организма.
Дефектный ген у больных ФКУ приводит к тому, что фенилаланин не идет по нормальной цепочке превращений. Он не становится тирозином, а тот не выполняет свои функциональные обязанности.
В итоге у больных аминокислота превращается в соединения, которых в организме не должно быть вовсе – фенилпировиноградная и фенилмолочная кислоты, фенилэтиламин и ортофенилацетат. Эти вещества копятся, словно мусор, и вызывают:
- Нарушение процессов жирового обмена в головном мозге;
- Недостаток нейромедиаторов, передающих нервные импульсы между клетками;
- Интоксикацию, которая отравляет ЦНС.
Это негативное воздействие значительно и необратимо снижает интеллект больного. У малышей быстро возникает олигофрения – умственная отсталость.
Симптомы
Клиника ФКУ имеет довольно яркое и специфическое проявление. Она заметна уже в первый год жизни ребенка. Основными симптомами являются:
- Вялость и незаинтересованность в окружающем мире;
- Повышенная плаксивость;
- Расстройство тонуса мышц;
- Судороги;
- Сниженная пигментация кожных покровов, волос и радужки глаз;
- Склеродермия;
- Эпилептические припадки;
- «Мышиный» запах мочи.
Однако главным признаком является нарушение в психоречевом развитии и микроцефалия. Это не означает, что заболевание проявит себя сразу же. Фенилкетонурия у новорожденных может не давать внешне заметных симптомов.
Определить проблему «налицо» становится возможным лишь на 2-6 месяц. Физически больные младенцы меньше отличаются от сверстников, нежели чем в психическом плане. Нередко головка ребенка меньше, чем положено его возрасту.
Зубки у таких деток прорезываются позже, сидеть и ходить они тоже не начинают в срок. Внешность ребенка с ФКУ характерна – это бледная белая кожа и светлые волосы с глазами. Это объясняется нарушением пигментации, вызванной отсутствием тирозина. Такая кожа чувствительна и склонна к появлению высыпаний под воздействием ультрафиолетового излучения.Дети в возрасте года не могут голосом показывать эмоции и понимать чужую речь. Их мимика невыразительна. С возрастом симптомы увеличиваются, словно снежный ком. Наблюдаются нарушения в поведении таких деток, психическая и умственная отсталость. Без лечения клинические проявления будут лишь прогрессировать.
Диагностика ФКУ
Достоверным способом установления наличия заболевания является медико-генетическая экспертиза. Сегодня она носит характер неонатального скрининга для массового обследования новорожденных. Такая процедура эффективна для выявления самых распространенных наследственных патологий. В это число входит и фенилкетонурия.
К дополнительному обследованию, направленному на точное установление болезни, относится биохимический анализ крови и общий мочи. ФКУ подтверждается в случае:
- Повышенного уровня фенилаланина, больше 900 мкмоль/л;
- Нахождения фенилпировиноградной, фенилмолочной или фенилуксусной кислот;
- Положительной пробы Феллинга.
Лечение
Каким бы опасным ни было заболевание, с ним можно бороться, сохранив нормальное качество жизни. Главной линией в лечении ФКУ является диетотерапия. Помимо этого, внимания заслуживают:
- Энзимотерапия фенилаланингидроксилазой, фенилаланинаммониалиазой;
- Прием тетрагидробиоптерина – «Сапроптерин»;
- Метод больших нейтральных аминокислот.
Эффективно показало себя применение пищевых гликомакропептидов. Они снижают концентрацию фенилаланина в крови больного и его головном мозге.
Такая терапия направляется на адекватное физическое развитие детей. Создан экспериментальный метод лечения ФКУ.
Его смысл состоит в непосредственном введении гена фенилаланингидроксилазы в поврежденные клетки печени. В России данные методики еще не нашли себе применения.
Прогноз и профилактика
Жизнь человека с ФКУ, напрямую зависит от своевременности начала лечения. Если помощь была оказана, то длительность жизни больных и ее качество такое же, как у здоровых людей.
При развитии симптоматической картины, длительность жизни значительно сокращается. ФКУ может стать причиной инвалидности, в случае развития необратимых нарушений ЦНС.
Следовательно, прогноз напрямую связан с формой заболевания и своевременностью лечения.
Особой профилактики для предотвращения фенилкетонурии не было разработано. Однако определенные меры могут предотвратить патологический процесс:
- Генетическая консультация необходима для людей, которые планируют зачать ребенка. Можно заранее выявить носительство гена ФКУ и других наследственных патологий. Оценить риск рождения детей с данными заболеваниями. Таким образом, родители будут заранее готовы к возможным рискам. И при рождении больного малыша, смогут сразу начать лечение, сохранив его здоровье;
- Скрининг для новорожденных, то есть массовое обследование по выявлению наиболее часто встречающихся генетических патологий. Это позволяет поймать фенилкетонурию на ранней стадии, следовательно, оказать своевременную помощь;
- Консультация для женщин, больных ФКУ. Это позволит им спланировать беременность и верно питаться во время вынашивания плода.
Продукты для детей с ФКУ
Для ребенка, который болен фенилкетонурией, очень важно правильное питание. Родители должны знать какие продукты больному можно употреблять в пищу. Чтобы избежать токсического действия фенилаланина, необходимо исключить из питания животный белок. Чем раньше это сделано, тем выше вероятность сохранения нормальной нервной системы.
Однако полное ограничение белка невозможно. Ведь это важное условие для адекватного физического и умственного развития.
Поэтому больные дети получают лекарственные заменители, обычно в виде специальных порошков.
Они содержат все необходимые питательные вещества, которые имеются и в обычных белковых продуктах, но не опасны для больных. Для грудничков показано применение особых смесей – «Афенилак» или «Мдмил-ФКУ-0».Хотя мать и может кормить грудью сама, она должна придерживаться особой диеты.
Для малышей разного возраста используют специальные продукты для восполнения белковых запасов – «Берлафен» и «Циморган», «Минафен» и «Апонти». Дети постарше не должны иметь в меню белковой пищи.
По протоколу существует таблица, в которой продукты питания для них разделяют на 3 списка в зависимости от содержания фенилаланина:
- Красный – нельзя употреблять (мясо и колбаса, рыба и яйца, сыр, творог, орехи, хлеб, кондитерские изделия);
- Оранжевый – возможно небольшое количество в рационе (молочные продукты, рис и кукуруза, картофель и капуста);
- Зеленый – пища, на которую не накладываются никакие ограничения (ягоды и фрукты, зелень, крахмал, мед, сливочные и растительные масла, рисовая и кукурузная мука).
Существуют специальные низкобелковые продукты, которые позволяют больным питаться более полноценно. Родителям, а в будущем и самому ребенку, важно уметь рассчитывать количество фенилаланина в пище.
Диета
Правильно сформированный рацион, ограничивающий белок, является основой лечения ФКУ. Цель – соблюдение соответствующего возрасту уровня фенилаланина. Он должен быть:
- Для грудничков 120–240 мкмоль/л;
- Для дошкольников не больше 360 мкмоль/л;
- Для школьников не выше 480 мкмоль/л;
- Для старшеклассников до 600 мкмоль/л.
Чтобы облегчить родителям больного ребенка расчеты, 1 г белка условно принимают за 50 мг фенилаланина. Даже при соблюдении диетотерапии, пациенты с ФКУ должны дополнительно принимать витамины, особенно группы В, и минеральные соединения, богатые кальцием и фосфором, железом и важными микроэлементами.
А также рекомендовано применение препаратов, содержащих карнитин. Придерживаться диеты, безусловно, следует как можно дольше.
С возрастом допустимо расширение продуктов питания, так как толерантность, то есть терпимость к фенилаланину, у больных растет с годами. Постепенно в рацион вводят небольшое число круп и молоко.
Одновременно ведут контроль за состоянием посредством электроэнцефалографии и биохимических анализов крови.
И хотя после 18 лет показано расширение диеты, в будущем больные должны придерживаться рекомендации воздержания от животных белков. Особенно строгой является диета у девочек, так как они будущие матери и их главная задача родить здоровых детей.
Врач невролог высшей категории Шенюк Татьяна Михайловна.
Источник: http://umozg.ru/zabolevanie/fenilketonuriya-u-detej.html
Фенилкетонурия
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.
; встречается с частотой 1 случай на 10 000 новорожденных. В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка.
Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1).
Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше.
При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты.
Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов.
В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина.
Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов — фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев.
С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома.
Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко — врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами.
К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы.
Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет.
При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным.
Скрининг-тест проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза).
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси — Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет — Максамум-ХР и др.
Основу диеты составляют низкобелковые продукты — фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям — ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Прогноз и профилактика фенилкетонурии
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием.
Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода.Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Источник: https://www.krasotaimedicina.ru/diseases/children/phenylketonuria
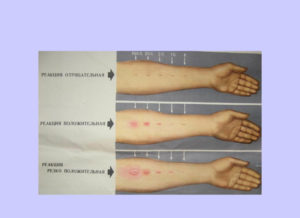
Диаскинтест положительный результат у ребенка – что делать

Диаскинтест положительный у ребенка бывает редко. Такой результат иногда приводит маму к отчаянию, шоку. По меньшей мере, родители очень расстраиваются, узнав о нехорошей реакции диаскинтеста у своего малыша. Положительная реакция Манту встречается чаще. Но огорчений родителям приносит меньше.
Эмоции мамы понятны. Потому что это значит диагноз туберкулез. Теперь надо как-то жить дальше. Причем, как то менять свою жизнь для того, чтобы избавить ребенка от неприятного инфицирования.
Что делать
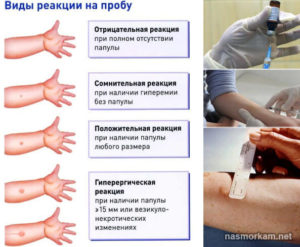
Сначала маме надо успокоиться. Истерия на результат никак не повлияет – однозначно. Во-первых, надо убедиться, что у вашего ребенка положительный диаскинтест, а не прба Манту. Не надо их путать. Мантушка ставится на одной руке.
Она может дать реакцию как на туберкулез, так на прививку от него (БЦЖ). А диаскинтест ставится точно так же, только на другой руке. Техника постановки, оценка результатов – все одинаково. Отличается только вводимым аллергеном (препаратом).
Конкретно, диаскинтест становится положительный, если организм инфицирован туберкулезом.
На какой руке что поставлено – это все записывается в карточке ребенка. Если вы не уверены, на какую именно пробу у вас положительная реакция – уточните. Выяснить вопрос, на какую руку вам ставили диаскинтест, можно у участкового педиатра, процедурной медсестры (которая вам ставила этот укол) или у врача фтизиатра.
При положительном результате диаскинтеста
Если вы все-таки услышали то, чего так боялись услышать – приготовьтесь ближайшее время посвятить на обследование ребенка и своей семьи. Все взрослые должны пройти флюорографию. Малыш – кроме стандартных анализов, еще УЗИ и томограмму легких (послойные рентген снимки).
Со всем этим добром вы идете к фтизиатру на прием, прихватив для осмотра ребенка. Не забывайте вести себя спокойно, чтобы не напугать дитя. Внушите ему, что ничего страшного с ним не будет.
Все будет хорошо! В крайнем случае, придется попить таблетки – а это не смертельно, даже не больно.
Если все анализы хорошие
Но мама не должна расслабляться, если все пройденные анализы нормальные, а диаскинтест положительный.
Такая картина может быть потому, что от момента внедрения до выявления микобактерий или клинических изменений должно пойти продолжительное время.
То есть, ребенок может быть инфицирован туберкулезной инфекцией, просто выявить ее очень трудно. Иногда положительный диаскинтест может быть единственным признаком начинающегося туберкулеза.
Из моей практики: в детском туберкулезном санатории долго лечилась девочка Соня. Она была из благополучной, полной семьи. Ее папа работал на скорой помощи. Сонечка лечилась около года. На протяжении всего времени лечения, у нее брали всяческие анализы, но они были «хорошие».Положительными были лишь проба Манту и диаскинтест. Соня получала противотуберкулезное лечение. Врачи долго не могли определить, какой орган девочки страдает от этой инфекции. Ведь, туберкулез может обосноваться где угодно, а не только в легких. К концу лечения у Сони обнаружили кальцинаты в печени.
Это значит, что туберкулез был там и уже излечился.
Что делать дальше
Важно не запустить, остановить рост микобактерий сразу. Поэтому, если результат диаскинтеста положительный, назначают противотуберкулезные препараты. Их нужно принимать без перерыва. Иначе разовьется лекарственная устойчивость. Тогда микобактерии, вызвавшие туберкулез у вашего ребенка, привыкнут к этим таблеткам.
И вылечить инфекцию будет сложно, потому что противотуберкулезных препаратов всего очень мало. Обычно их назначают комплексно, чтобы вывести микобактерию из организма совсем, пока она не приспособилась к лекарству. Поэтому к лечению надо подходить со всей ответственностью и серьезностью.
Лекарства принимать по правилам, о которых информирует фтизиатрическая служба.
Если есть сомнения по поводу туберкулеза
Чтобы убедиться окончательно – есть ли туберкулез или нет – просите у врача направление на ПЦР диагностику. Этот окончательный анализ «поставит все точки над I».
ПЦР анализ платный, система ОМС его пока не финансирует. Это исследование на уровне генных изменений при инфекциях, которые трудно выявить. На сегодняшний день ПЦР диагностика является самым точным результатом на туберкулез.
Если туберкулез у ребенка подтвердился
Есть несколько вариантов лечения. Его назначает врач фтизиатр, руководствуясь стадией процесса. Если все запущено – отправляют в стационар.
Если не все так страшно, то ребенку могут предложить санаторное пребывание с круглосуточным или дневным медицинским наблюдением, комплексным лечением, усиленным питанием.
Так же существуют специальные детские садики, которые отличаются от обычных только тем, что дети там, плюс ко всему, получают противотуберкулезное лечение. Некоторые детки лечатся дома и никуда не ходят.
Если у вас еще есть вопросы по поводу положительного диаскинтеста – пишите. Отвечу, насколько мне позволяет моя компетенция.
Источник: https://detimy.ru/diaskintest-polozhitelnyiy-rezultat-u-rebenka-chto-delat/

